AEZS Transformation zum Big Player
So hat man schon Produzenten für Zoptrex und Macrilen gefunden und man kümmert sich allgemein um die Verkaufsstruktur.
Es wird keine Zeit vergeudet. Man kümmert sich schon um die Vermarktung, bevor die Medikamente eine zulassung bekommen.
Ob das darauf deutet, dass die Zoptrex ergebnisse gut sind kann man schwer beantworten.
Aber man merkt schon, das man bei einer Zulassung von Zoptrex einen konkreten Vermarktungsplan hat. So wird Zoptrex für Gebärmutterhalskrebs nur von AEZS vermarktet. Aber für die anderen Indikatoren, also anderen Krebssorten wird für ein Partner gesucht.
Weil sie selbst wissen, das sie nicht genug Geld haben um diese alleine auch zu vermarkten.
Dies zu wissen ist wichtig für unsere Risikoanalyse,step by step auch kleinste Infos können uns hier
sicherer schlafen lassen und uns vor Fehlentscheidungen schützen.
Einige Pharmakontakte habe ich und suche nach Anhaltspunkten um diese zielgerichtet
zu kontaktieren.
Sowohl Zoptrex als auch Macrilen werden jetzt schon von externen Firmen produziert. Jetzt wird nach je einem zweiten Hersteller gesucht um nicht abhängig zu sein:
In addition, we expect to incur costs related to the validation of a second supplier for both products to be able to fulfill the expected demand. (letzte CC)
Zu den bisherigen hat Dodd nur von "größeren Herstellern in Europa und Asien" erzählt.
In der alten Macrilen Studie wurde bei den Probanden ein cut-off-point von 2,7 ng/ml ermittelt . In der neuen Phase 3 Studie zu Mac wurde der cut-off-point von 2,8 ng/ml gewählt. Also daher praktisch derselbe.
Ich hatte mir mal selbst die Frage gestellt, wie will die FDA den cut-off-point von Macrilen eigentlich wissen. Denn die FDA ist eine Zulassungsbehörde, die Daten auswertet und dann eine Zulassung erteilt oder nicht. Sie hat an der Forschung an Macrilen nichts zu tun. Sie können nur anhand der Daten die Forschung beurteilen.
Dadurch kam ich zu den Urteil, dass die FDA den cut-off-ponit nur anhand der alten Phase 3 Studie zu Mac beurteilen konnte. Dadurch lässt sich auch der cut-off-point von 2,8 ng/ ml erklären die die FDA ausgewählt hat. Genau derselbe, wie in der vorherigen Phase 3 zu Mac.
Aber wie wir alle wissen hatte die alte Phase 3 Studie Probleme. So war argine + gherlin während der Phase 3 Studie nicht mehr verfügbar. Dies führte auch zu Problemen bei Bedtimmung des cut-off-ponits.
Ausschnitt der alten Phase 3, welche die Autoren selbst die Probleme der Bestimmung des cut-off-points erwähnen:
Es gab einen Unterschied in den Schnittpunkten für Macimorelin-stimulierte Peak-GH-Werte und für diejenigen Kontrollen, die beide Tests gegen die gesamte Kohorte erhielten. Mehrere Faktoren könnten zu diesem Unterschied beigetragen haben, einschließlich der Zufallsunterschiede aufgrund der kleinen Stichprobengröße der ersten Kohorte (n = 10). Die Verwendung dieses Mittels zur Diskriminierung dieser Patienten mit Hypothalamuserkrankungen von Hypophysenkrankheiten, einschließlich Patienten mit neueren Bestrahlung oder Tumoren im Hypothalamus wie Hamatoomen, bleibt abzuwarten. Arginin + GHRH ist unter diesen Umständen mit falsch-normalen Reaktionen verbunden. Es kann sich herausstellen, dass Patienten mit hypothalamischen Patienten nicht auf diesen Agenten reagieren und ein wichtiges diskriminierendes Werkzeug liefern. Weitere Studien mit einer größeren Stichprobengröße wären erforderlich, um diese Frage endgültig zu beantworten, obwohl dies nur möglich wäre, wenn Geref Diagnostic (Serono) wieder verfügbar wird. Unangenehmer Geschmack war das einzige unerwünschte Ereignis, das häufig mit Macimorelin gemeldet wurde. Alternative Strategien zur Verbesserung der Schmackhaftigkeit einschließlich der Verwendung eines Verdünnungsmittels außer Wasser werden in laufenden Versuchen getestet.
Dadurch kann man auch verstehen, weshalb Dodd die FDA gefragt hat welchen cut-off-ponit sie für die die neue Macrilen Studie nehmen sollen.
Da die richtige Bestimmung des cut-off-ponits in der alten Studie zu Macrilen nicht richtig erschien. Da die Stichprobengröße von 10 Leuten, wegen dem fehlen von argine+ ghrelin viel zu gering ist und kaum aussagekräftig für eine größere Gruppe ist . Außerdem gab es bei dieser kleinen Gruppe Abweichungen bei einigen Probanden bei der Diagonse von AGHD mit Macrilen und argine+ghrelin.
Daher konnte AEZS kaum den alten cut-off-ponit von der alten Studie nehmen und hat daher die FDA nach der Auswahl des cut-off-points gefragt. Aber die FDA konnte nur anhand der Daten der alten Phase 3 den cut-off-point für die neue Phase 3 beurteilen und dieser war der gleiche wie in der alten Phase 3.
Dadurch lässt sich auch erklären, weshalb einer der sekundärziele in der jetzigen Studie zu Mac die erneute Bestimmung des cut-off-ponit war.
Mit diesem wissen könnte man davon ausgehen, dass die FDA AEZS wegen der Erteilung der Macrilen Zulassung entgegen kommt. Da die FDA auch nicht ganz unbeteiligt war am Mac fail.
Die Daten der jetzigen Phase 3 Studie zu Macrilen waren auch sehr gut und man hat sich auch ans SPA-Protokoll gehalten. Ergomed hatte auch die Phase 3 Studie durchgeführt. Außerdem war die zusätzliche Studie zur Reproduzierbarkeit der Macrilen Studie auch sehr gut. Dadurch alleine würde es keinen sinn machen eine vollständige Wiederholung der Phase 3 Studie zu Macrilen zu verlangen.
Daher gehe ich von einer Zulassung von Macrilen aus. Aber mit einer angehängten Phase 4 Studie, da ich glaube dass die FDA wegen dem neuen cut-off-points zur Diagonse sicher gehen will. Außerdem gehe ich lieber von diesem Szenario aus, weil wie ja alle wissen das die FDA streng ist und keinem etwas schenkt.
So könnten schon mal Einnahmen generiert werden während man den besten cut-off-Point ermittelt.
Bin daher zuversichtlich, das wir ein positives Feedback von der FDA bekommen.
Fand meinen oberen Post wichtig, da man dadurch Dodds Aussagen zum cut-off-ponit auch nachvollziehen kann.
Optionen
| Boardmail an "silverfreaky" |
Wertpapier: Cosciens Biopharma Corp |
Wir werden es nächste Woche sehen, wenn die News über das FDA-Gespräch veröffentlicht werden.
Waren glaube ich 150 - 200 Mio.
Da lohnt sich das auf alle Fälle ( losgelöst vom medizinischen Nutzen für die Patienten)
Optionen
| Boardmail an "Androlyt" |
Wertpapier: Cosciens Biopharma Corp |
Habe 17 Bio Werte im Depot bisher leicht im minus damit,ist normal,kann sich bei news zum positiven oder negativen ändern.Cerulean Pharma ist momentan der interessanteste Wert am 31 März wird etwas bekannt gegeben von der Conference C.Vermute es gibt ein ein enormen Kursanstieg,total spanend,die anderen Werte laufen seitwärts.ist jetzt keine Werbung,wenn man mit Cerulean Pharma noch schnelle hundert Prozent macht könnte man mit dem Gewinn in aeterna reingehen ohne sorge was Aetherna ende april für news bringt.
Nur so ein Gedanke von mir.
Optionen
| Boardmail an "Androlyt" |
Wertpapier: Cosciens Biopharma Corp |
Und laßt uns hier bitte bei der Sache bleiben, denn die dürfte weit spannender sein, als die (unnötigen) Spekulationen über längst verblichene andere Werte... Danke.
Ich habe einiges zusammengetragen an Informationen zur Phase 3, welche einiges an Recherche gekostet haben. Schreibe die erst jetzt, weil ich vorher nicht so viel Zeit für ausführliche Texte hatte.
Zur Studie und statistischen Plänen:
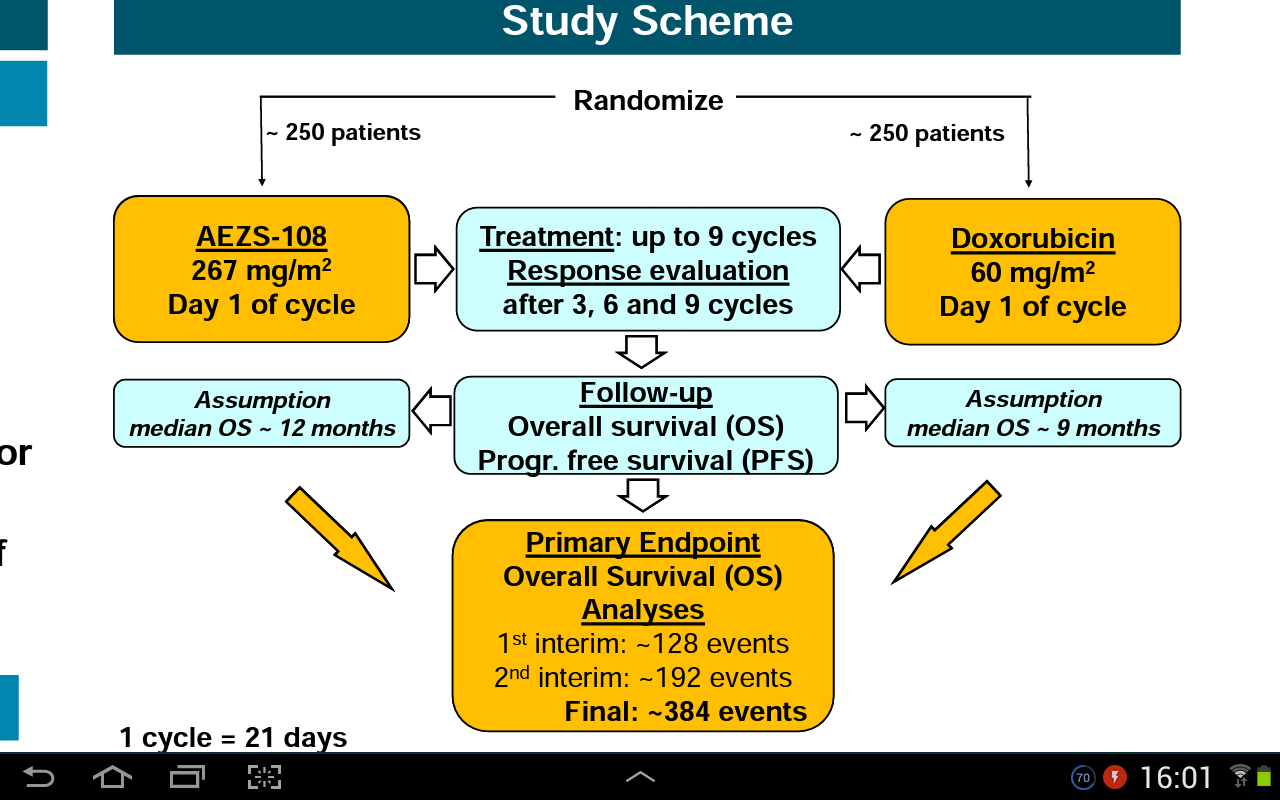
Open-Label, parallel Assignment, randomisierte kontrollierte Phase-III-Studie, um die Wirksamkeit und Sicherheit von AEZS-108 und Doxorubicin zu vergleichen.
Die Studie umfasst etwa 500 Patienten mit endometrialen Krebs vorbehandelt mit Platin-Taxan-Kombination.
Randomisierte Zuordnung zur Studienbehandlung (1: 1-Verhältnis):
- AEZS-108: 267 mg / m2 IV, Q3W, bis zu 9 Zyklen
- Doxorubicin: 60 mg / m2, IV, Q3W, bis zu 9 Zyklen
Response Auswertung jede 3 Zyklen; Frühere Neubewertungen zur Response-Bestätigung (RECIST) oder im Falle einer vermuteten Progression.
Patienten ohne PD (Pharmacodynamics) am Ende der Behandlung werden alle 12 Wochen bis zum Fortschritt neu beurteilt. Alle Patienten werden zum Überleben verfolgt.
Bei einer Teilstudie an ausgewählten Standorten wird die PK-Profile (Pharmacokinetics) von AEZS-108 und Doxorubicin bei insgesamt 40 Patienten untersuchen.
Primär Endpunkt (EP) ist Gesamtüberleben (OS).
Sekundäre EPs umfassen das progressionsfreie Überleben (PFS), die objektive Ansprechrate (ORR) und die klinische Nutzungsrate (CBR).
Die endgültige Analyse ist nach etwa 384 Todesfällen geplant; Die Stichprobengröße basiert auf der OS-Annahme von 12 vs 9 Monaten. 12 Monate OS Zoptrex und 9 Monate OS Doxorubicin,
Ein unabhängiger Data and Safety Monitoring Board (DSMB) überprüft die Sicherheitsdaten regelmäßig in Abständen von nicht mehr als 6 Monaten sowie die Ergebnisse von Zwischenanalysen, die nach etwa 128 Todesfällen (nur Nützlichkeit) und nach etwa 192 Todesfällen (Sicherheit und Wirksamkeit) geplant sind ).
Die Untergruppenanalyse ist geplant, um den potentiellen Wert des LHRH-Rezeptor-Expressionstests für die Vorhersage des therapeutischen Nutzens zu bewerten.
Angehängte Grafik:
screenshot_2017-03-27-16-01-59.png (verkleinert auf 39%)
screenshot_2017-03-27-16-01-59.png (verkleinert auf 39%)
Angehängte Grafik:
screenshot_2017-03-27-16-30-56.png (verkleinert auf 39%)
screenshot_2017-03-27-16-30-56.png (verkleinert auf 39%)
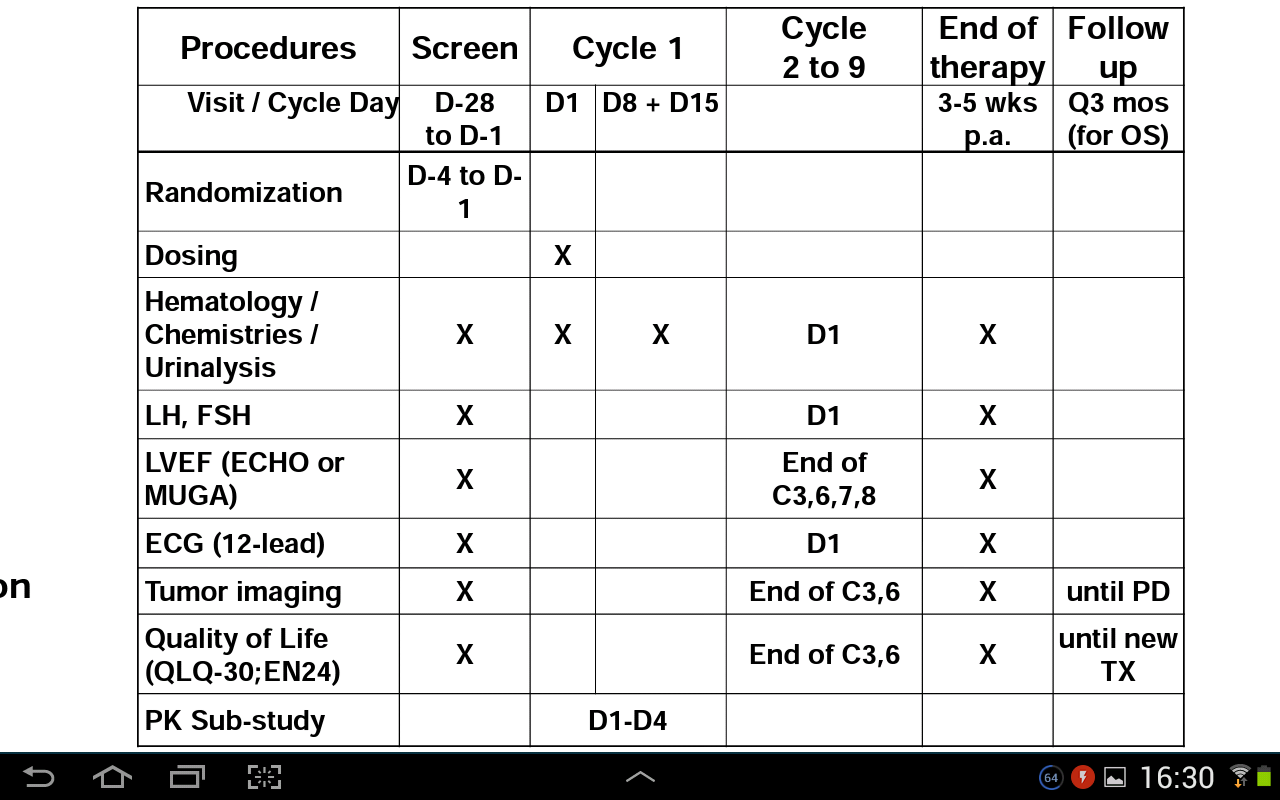
Unglaublich wichtig ist z.B. die Information, dass alle 6 Monate das DSMB die Patientendaten auf Sicherheit prüft. Unsere Studie ging knapp 4 Jahre, das bedeutet es gab für die Sicherheit der Studie mindestens 7 Überprüfungen durch das DSMB.
Das zeigt mir das Zoptrex viel sicherer ist, als Doxorubicin und ein geringeres Nebenwirkungsprofil hat als dox. Also sollten die sekundäre in der Phase 3 Studie gut vllt. sogar sehr gut sein im Vergleich zoptrex gegen dox.
Außerdem eine weitere wichtige Informationen ist, dass die Studie für 3 Jahre geplant wurde. Dabei ging man bei dox von einem Os von 9 Monaten und bei Zoptrex von einem OS von 12 Monaten aus, als man vom Studienzeitraum von 3 Jahr ausging.
Unsere Studie ging knapp 4 Jahre, also ein Jahr länger. Das bedeutet das es eine Abweichung im Os gab, also einen höheren Os als erwartet.
Anzumerken ist, dass das Os 3 Monate für Zoptrex niedriger in der Phase 3 bei der Plannung geschätzt wurde, als in der Phase 2 bestimmt wurde 15 Monate.
Die längere Laufzeit der Studie zeigt eine Verschiebung des Os allgemein.
Ich persönlich vermute, wie auch schon öfters diskutiert wurde, das Os bei der Dox und Zoptrex Gruppe höher lag, als ursprünglich vermutet bei der Erstellung der Studie.
Dox vermutlich ein Os von max 11-12 Monate, durch günstige Faktoren und guten Nachtherapien. Bei Zoptrex gehe ich auch von einem höheren Os von 12 Monaten aus, wie bei der Studienplanung vermutet wurde. Zoptrex OS wurde konservativ geschätzt nur 12 Monate statt 15 Monate wie in der Phase 2 . Zoptrex muss mindestens ein Os von 15 Monate in der Phase 3 Studie haben, sonst lässt sich einfach nicht erklären wie 120 Patientrn länger als 18 Monate überlebt haben. Daher verstärkt sich die Vermutung, dass diese Probanden zum großenteil wegen Zoptrex lãnger leben. Denn wenn es nicht so wäre, hätte das DSMB bei beiden internen Analysen auf Nutzen und Effektivität der Studie , die Phase 3 Studie zu Zoptrex abgebrochen. Ich sehe nicht, wie Dox bei einer anfänglichen Schätzung von 9 Monaten beim Studiendesign, 120 Patienten länger als 18 Monate leben kõnnen durch dox alleine. Sonst wäre auch die Studie abgebrochen worden und, die Studiendauer länger als 4 Jahre.
Fazit: ich gehe bei beiden Probandenarmen von einerr Verschiebung des Os aua, aber mehr zugunsten Zoptrex. Daher vermute ich , da ein OS unterschied von 3 Monaten zwischen Dox und Zoptrex mit einer hohen Wahrscheinlichkeit erfüllt werden sollte.
Alles nur meine Meinung.
Aber würde mich über Feedback bei meiner Überlegung freuen